Contact us
- No. 28, Lane 210, Doi Can Street, Doi Can Ward, Ba Dinh, Hanoi
- info@icharm.com.vn
- 0906.534.111
- 0943.839.666
Gallery


Share the next article about CRISPR/cas9 gene editing technology. If anyone has the same interest, let's discuss it together
After an introduction to the development history of the CRISPR/Cas9 system, today's article topic is how to create a virus capable of editing genes using CRISPR/Cas9 technology?
Figure 1 shows the results after cutting a targeted gene segment of lentiCRISPR/Cas9 on a cancerous human cell line. After using the CRISPR/Cas9 system to cut the target gene, each cell is isolated and grown into lines, separating the two DNA strands for sequencing. The results showed that cell line 1 was not cut, the gene segment at the leading position was completely similar to the natural body, while cell line 2 was the one in which both DNA strands were cut with different mutation results. cell line number 3 has only 1 cut chain, cell line number 4 is derived from 2 different original cells, so the sequencing results show that there are 3 mutant lines: 1 chain has lost 1 nucleotide ( nu), one chain lost 4 nu and had a point mutation replacing C with A, one chain lost 4 nu and had a point mutation replacing G with T. So how can we do this, I will show Briefly present the mechanism and process for you to understand.
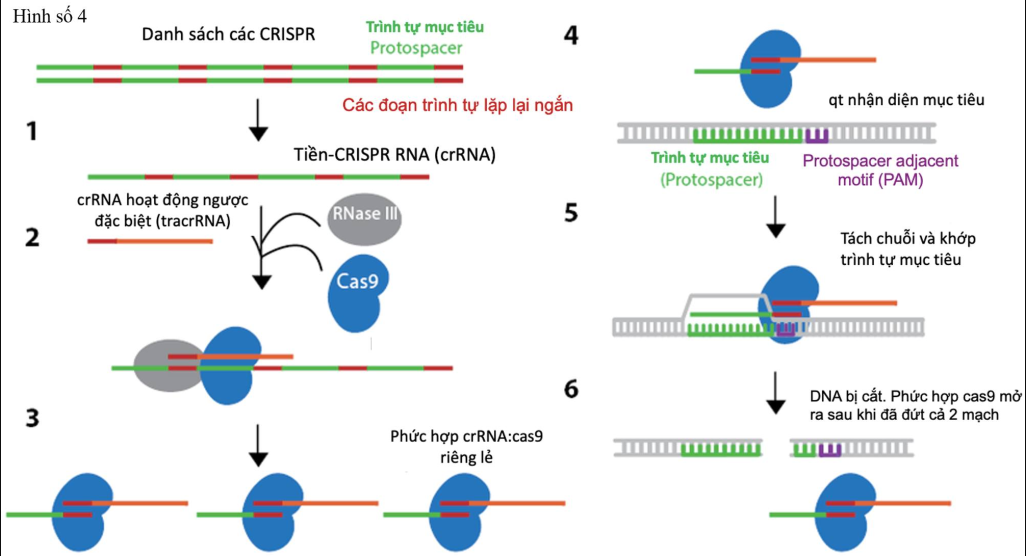
CRISPR/Cas groups have many different subclasses of Cas enzymes (see table 1). I only focus on researching and using a lot on Cas9. Other cases have various disadvantages and are currently being studied further to improve their capabilities.
I. Real-life visual example of the CRISPR/Cas9 process
That's short, but if we start working, at the fastest speed like nonstop, it would take at least 7 to 10 days to create a virus that can edit genes according to the mechanism of CRISPR/Cas9. . The steps to create a virus capable of gene repair are as follows:
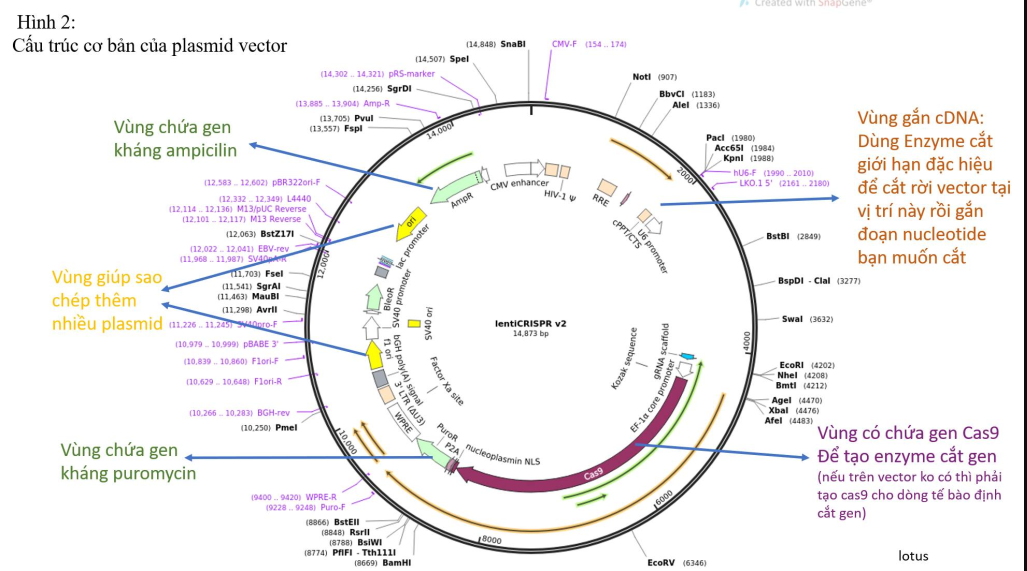
1. Select the vector plasmid, cut and purify the vector with a cutting enzyme specific to the vector you use. The basic vector structure is depicted in figure 2. Now we have a vector cut at a specific location and can attach cDNA fragments (you create cDNA from the desired oligo RNA, also known as guide RNA). (guide RNA). The site of the enzyme cut is important for placing the oligo, so that the cDNA can then be attached back to the cut site of the vector. That's why people who do it a lot can see it oligo sequence that identifies the type of plasmid being used.
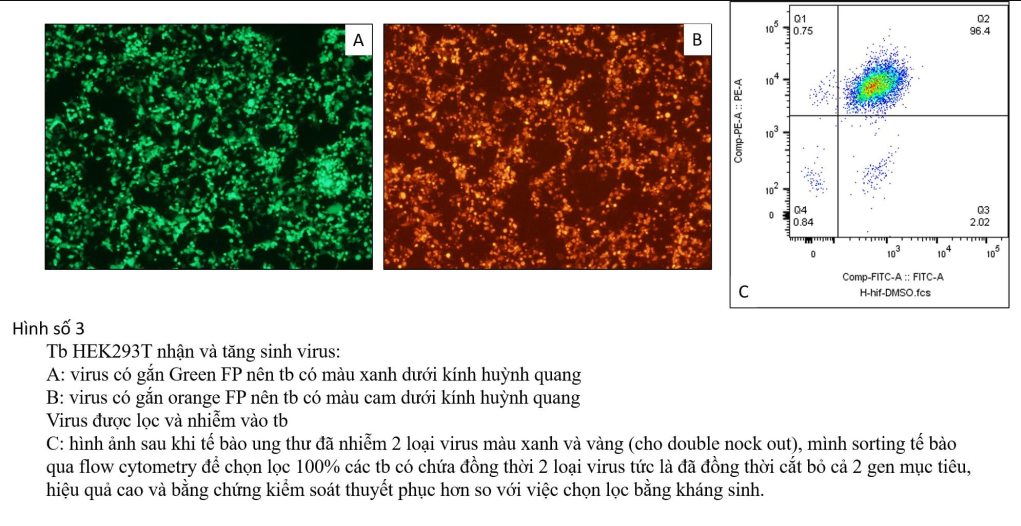
(Note: I often use plasmids with the characteristics I want so that I can later determine which cells have been infected with the virus and which cells have not been infected, because in reality when the virus invades The cell population depends greatly on the concentration of the virus you create, the reaction efficiency and also depends on the chemicals and processes. Therefore, to control the results of the experiment, I usually Choose plasmids with built-in colors like GFP, RFP... or pre-installed antibiotic resistance genes like puromycin or neomycin.... Thanks to these characteristics, after being infected with the virus, you can create lines that guarantee 90%-100% of cells. has been infected with a virus, meaning the target gene has been completely cut off.)
2. Order gRNA: you order this from the company, it's actually just Nucleotide sequences about 20-30 nucleotides long, usually very fast, it only takes 1 day for the gRNA set to arrive to you. But to set the gRNA standard, you need to understand the special features of the plasmids, cutting enzymes, and the gene sequence you want to cut, because genes are often very long but only have a few meaningful and truly important segments for the gene. Therefore, when that position is mutated or replaced by a nucleotide, the function of that gene will be lost. For example, I often choose the promoter region or exons from 1 to 10. And usually to delete a gene, I usually do it with 10 sgRNAs because you cannot guarantee that you choose 2 sg for both positions, because when editing the gene, it will be lost. the role of that gene or not.
(This is a bit confusing, isn't it? That is, even if you have successfully cut that gene segment out of the cell's gene, if that gene segment is not important for that gene's role, that gene will still function normally. Transcribe, translate and create normal proteins. It's like having a cancer-causing mutation that has no effect on the body. Therefore, the gene is selected to completely inactivate the gene You also need experience or you can look in the articles and maybe they will give you information about the procedures they have succeeded in.)
3. Connect the cDNA to the plasmid: use ligation mix reagent to connect the sgRNAs (forward and reverse) to the plasmid. At that time, the plasmid will close itself.
4. Transformation
Transfer the plasmid into bacteria such as E.Coli. I often use DH5a
Then inoculate the bacteria into the agar plate. After 12 hours you will have VKs growing, in the VKs there will be the plasmid you want
5. Cultivate VK to increase the amount of plasmid: take a colony that is not too big, not too small, do not grow together with other colonies to increase the possibility of catching a colony with a plasmid containing the target gene. (This part is like choosing a mate. If you don't choose well, you've had enough, ha ha) and then culture it in LB solution to let the bacteria multiply.
6. Plamid separation and purification: by kit only. Fast and highly effective
7. Create Virus: